No results found
Loading...

Over the past several years, proteomics has seen a surge in interest among life scientists and the biopharma industry, akin to the previous rise of genomics. It is becoming more evident that uncovering critical insights into the proteome can provide a wealth of knowledge into disease and physiology and thus become a new avenue to unveil novel targets for drug discovery and new molecular signatures for biomarker development.
Although the goal of comprehensively surveying the proteome landscape remains elusive, recent advancements have brought us closer to achieving it. Today, we can dive deeper with advances in single-cell technologies or tap into near-comprehensive proteomics to study thousands of proteins in a given sample. Scientists can study the proteome faster and more efficiently than ever thanks to throughput, sensitivity, and cost improvements both in mass spectrometry (MS) and in supporting technologies for sample preparation.
Proteomic approaches may be divided broadly into two types of strategies: targeted and untargeted. Targeted or affinity-based methods frequently require a bait, such as an antibody or aptamer, to facilitate binding to a specific target. In targeted MS approaches, specialized software incorporating preselected protein or peptide lists is required. While exact, this approach allows researchers to evaluate only particular proteins of interest.
On the other hand, unbiased MS-based analysis strategies provide insights into the overall composition of samples without relying on specific baits. In that regard, MS offers the potential to detect a wide range of proteins within a sample. Unlike affinity-based approaches, which often require extensive sample pretreatment, unbiased MS is not limited to a predefined set of proteins and is higher throughput due to reduced sample preparation demands.
MS has greatly contributed to proteomics due to the instrumentation’s ability to comprehensively identify proteins and peptides with high sensitivity and resolution. This was not always evident, as many researchers shied away from studying the proteome with MS due to the belief that MS was too complex, unaffordable for most researchers, and limited to providing low proteome coverage. These concepts have not entirely disappeared, but they are significantly less prevalent. Today, many MS-based technologies have matured and are now widely available. Advanced capabilities are driving adoption not only because of increased speed, lower costs, and ease of use, but also because of performance improvements that can help researchers achieve greater proteome depth more quickly.
Sample multiplexing, via techniques such as isobaric labeling with tandem mass tags (TMTs), has emerged as a pivotal technique in MS-based proteomics because it enables multiple samples to be analyzed simultaneously in a single MS experiment. The TMT approach currently allows the multiplexing of 18 samples (thereby enhancing throughput and reducing experimental variability).
In the context of TMT, each sample is labeled with a unique tag (which essentially barcodes the peptides) that fragments during tandem mass spectrometry to produce reporter ions. These ions are then used to quantify the peptides and, by extension, the proteins from which they originated. The use of TMT labeling facilitates the comparison of protein expression across different samples, making it an invaluable tool for proteomic studies. However, sample preparation for such a strategy is extensive. It requires proteolytic digestion, labeling, pooling, and final desalting. In addition, multiple samples must be prepared in parallel. That said, sample multiplexing ultimately enables researchers to delve deeper into the proteome, thereby expediting discoveries with greater efficiency.
Along with advancements in sample processing, mass spectrometers have become more sensitive, accurate, robust, and amenable to higher throughput data analysis. For example, the Orbitrap Astral mass spectrometer is a groundbreaking instrument in the field of proteomics, significantly enhancing throughput capabilities. This high-resolution mass spectrometer has exceptional speed and sensitivity, enabling the rapid and accurate identification and quantification of proteins. Such benefits are enhanced when using sample multiplexing, thereby increasing throughput and reducing experimental variability. Furthermore, its superior resolution and mass accuracy facilitate the detection of low-abundance proteins, enhancing the depth of proteome coverage.
The increased throughput provided by the modern-day mass spectrometers not only accelerates the pace of research but also contributes to more robust and reproducible results, thereby driving forward the field of proteomics. These advancements have made MS faster and more cost-effective, akin to next-generation sequencing and its broad usability. Today, the time required to collect proteomics data for near-complete proteomes has drastically decreased from days to hours.
While MS has advanced tremendously in recent years, becoming more user-friendly, standardized, and accessible, a significant challenge for the industry remains the upstream bottleneck of sample preparation.
A crucial component for using MS-based proteomics is reliably, reproducibly, and rapidly preparing samples to ensure they are suitable for MS-based analysis. While standard sample preparation techniques have been adopted by the proteomics community, sample preparation for MS-based proteomics is still a multifaceted, multistep process that demands flexibility to support a range of workflows based on the type of proteomics study being performed.
Sample preparation for MS-based proteomics often includes processes such as sample lysis, prefractionation, depletion or enrichment of proteins, enzymatic digestion, cleanup, tagging, or concentration, depending on the specific requirements of the sample and targeted workflow. Examples of different manual and automated proteomics sample prep techniques are shown below (Figures 1 & 2).
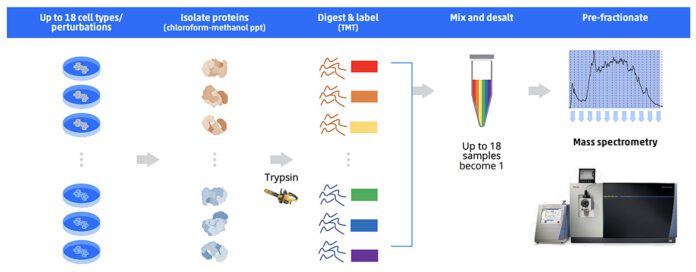
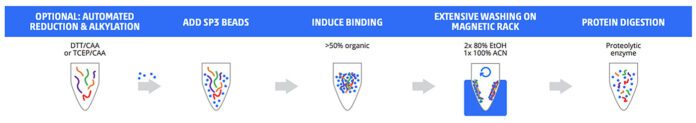
To that end, scientists have expressed concern that the pace of upstream sample processing is not commensurate with the speed of MS data acquisition. Thus, upstream sample processing, in many cases, now represents a bottleneck for many proteomics researchers.
Sample processing automation with robotics in tandem with MS is one area that can address the challenge of upstream processing. This approach has several benefits, including:
Many laboratories have incorporated automated sample preparation solutions into proteomic studies with great success. For example, scientists from Greifswald Medical School developed a web-based application for MS studies on Opentrons laboratory robotics for protein research. In a recent article, the scientists reported that the end-to-end automated application offered flexibility, reduced hands-on time, and was higher throughput compared to standard manual sample prep, which involves 21 pipetting steps per sample, and over 2,000 pipetting steps for 96 samples (Reder et al. Proteomics 2023 Sept. 29; doi: 10.1002/pmic.202300294).
Another study from the University of Copenhagen used automated robotics from Opentrons to process 192 samples in 6 hours for clinical proteomics. The setup, which enabled complete automation of proteome and phosphoproteome sample preparation in one step, was used to investigate the impact of cancer immunotherapy on the plasma proteome of patients with metastatic melanoma. In a recent preprint, the study’s authors wrote, “[This] automation strategy enables protein digestion and loading of almost the entire resulting peptide sample, which greatly increases efficiency while reducing the cost of processing” (Kverneland et al. bioRxiv 2023 Dec. 22; doi: 0.1101/2023.12.22.573056).
Making sample preparation protocols widely available throughout the life sciences community is crucial for the broader adoption of laboratory automation. Automated workflows should be standardized and open source. One such workflow was developed recently at a Harvard Medical School laboratory led by one of this article’s co-authors (Paulo). This workflow proved its versatility for streamlining proteomic profiles in yeast studies (Liu et al. J. Am. Soc. Mass Spectrom. 2021 May 5; 32(6): 1519–1529). Openly available protocols can dramatically expand the usage of automated robotics, especially when large-scale proteomic studies are necessary.
Recent advances in MS technology have taken proteomics from a niche to a partner for genomics. A slow and costly process has transitioned into a broadly available tool, similar to the trajectory seen with next-generation sequencing, and is now allowing scientists to study thousands of proteins across multiple samples. These new MS advancements empower researchers to delve deeper into the proteome, enabling them to scale throughput and detect approximately 14,000 proteins, thereby expediting discoveries with greater efficiency.
However, this new and improved accelerated pace of MS accessibility has also imposed significant pressure on the front-end sample processing. The manual processing and the liquid handling of samples are inherently low throughput, can introduce human error, and lead to increases in inconsistency. This is precisely the point at which the application of laboratory robotics and automation becomes indispensable. By improving turnaround time and reducing costs, automation is poised to accelerate the pace of discoveries in proteomics by enhancing efficiency, reducing upstream bottlenecks, and expanding the scope of research.
Automation also improves precision in handling individual cells and allows for the analysis of larger and more complex experiments (by processing multiple samples to capture results corresponding to various time points or drug doses). The expansion in throughput paves the way for broader applications in proteomics, opening doors for more accurate diagnoses and drug development opportunities.
Over the next decade, the integration of automation into the proteomics field will continue to become indespensible, providing scientists with a means to explore further the proteome and offering biopharma and clinical stakeholders novel insights into disease. However, the industry needs to continue to invest in tools and techniques that will allow researchers to scale their endeavors, focusing on scientific breakthroughs rather than manual tasks at the benchside.
James Atwood, PhD, is general manager of robotics at Opentrons Labworks, and Joao A. Paulo, PhD, is instructor in cell biology and co-director of the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School.































